Mugurkaula amiotrofijas apraksts 1. tipa bērnu veselības stāvoklim. Mugurkaula muskuļu atrofija (SMA) bērniem. Iemesli. Kas izraisa SMA
Ar SMN saistītā MCA vai 5qSMA vai proksimālā MCA parasti tiek iedalīta trīs kategorijās. Parasti var atšķirt arī CMA0 (nulle) un CMA4. Tādējādi ir vairāki galvenie SMA veidi, un tie visi darbojas dažādos veidos. Process var attīstīties dažādos dzīves periodos, tam ir savas klīniskās iezīmes, savs kursa raksturs, prognoze un nepieciešamais palīdzības un atbalsta apjoms.
1. tips- visgrūtākais ar ātrāko debiju, 3. tips- vismazāk smags, ar novēlotu sākumu. Daži eksperti arī izšķir 4. tipu vidēji smagai vai vieglai SMA ar pieaugušo sākumu.
Slimības specifika ir tāda, ka katram bērnam, pat vienas grupas ietvaros, SMA norit atšķirīgi, individuāli. Ārēji tas izpaužas iespējamā kustību diapazonā - daži bērni spēj turēt galvu, nedaudz pacelt rokas un pacelt kājas, bet citi vienkārši guļ klasiskajā vardes pozā un var tikai nedaudz pakustināt kājas un pirkstus.
Viktors Dubovits, viens no vadošajiem skaitļiem SMA vadības jomā 90. gados, ieteica papildus parastajai klasifikācijai izmantot arī sarežģītāku skalu. Piemēram, ir CMA1, un tā apakštipi būs 1.1, 1.2, 1.3, t.i. no 1.1 līdz 1.9. Šo shēmu izmanto Itālijā.
Amerikas sistēma balstās uz ABC skalu, kur B ir klasisks, A ir vājš tips un attiecīgi C ir spēcīgāks. Sistēma ar ABC apakštipiem skaidri raksturo SMA klīniku un ļauj jums izvēlēties atbalstošu terapiju bērnam atkarībā no apakšgrupas un atbilstošās prognozes. Šī sistēma tiek izmantota Krievijā un citās valstīs.
Svarīgs! Ģenētiskais tests nevar noteikt SMA tipu. Veids tiek noteikts, pamatojoties uz bērna funkcionalitāti.
CMA0
Slimības simptomi izpaužas dzemdē, ja auglim nav motora aktivitātes. Kopš dzimšanas bērns ir izteicis vispārēju muskuļu hipotoniju ar raksturīgu "vardes" pozu, un spontāna motora aktivitāte tiek strauji samazināta. Cīpslas refleksi netiek iedarbināti.
Parasti šiem bērniem ilgu laiku seko pediatri un neirologi ar perinatālās encefalopātijas diagnozi. Dažreiz ārsti gausa bērna simptomu kompleksu saista ar grūtām dzemdībām. Bet visi bērni ar perinatālo encefalopātiju un ar sarežģītas dzemdības sekām pietiekami ātri un labi pielāgojas, pakāpeniski uzlabojas, atšķirībā no bērniem ar SMA.
Prognoze ir ārkārtīgi nelabvēlīga - bērni mirst, parasti, ļoti agrīnā vecumā(līdz sešiem mēnešiem) no starplaikā esošām slimībām (sarežģī pamatslimības gaitu).
Parasti CMA0 un CMA1 tiek apvienoti.
CMA1
Ar I tipa mugurkaula muskuļu atrofiju (Werdnig-Hoffmann tips) māte jau grūtniecības laikā var pievērst uzmanību vēlīnai un vājajai augļa kustībai. Kopš dzimšanas bērnam ir plaši samazināts muskuļu tonuss (“ļengana bērna” sindroms). Kopš pirmajiem dzīves mēnešiem augšējā un. Muskuļa vājums un atrofija apakšējās ekstremitātes, kam seko stumbra un kakla muskuļu iesaistīšanās. Šīs muskuļu izmaiņas bērnus nespēj sēdēt. Muskuļu atrofiju un muskuļu šķiedru raustīšanos parasti maskē labi izteikti zemādas tauki. Raksturīgs simptoms ir izstieptu rokturu pirkstu neliels trīce (trīce). Dažreiz tiek konstatēta mēles muskuļu raustīšanās.
Tipiska pazīme ir arī cīpslu refleksu (ceļgala, Ahileja) pavājināšanās vai pilnīga izzušana, locītavu normālas kustības ierobežošana un skeleta deformācijas. Starpribu muskuļu vājuma dēļ bērna krūtis kļūst saplacināta. Tā kā muskuļu vājuma rezultātā pietiekama plaušu ventilācija nenotiek, pievienojas biežas elpceļu infekcijas, dažādas elpošanas traucējumi... Bērnu garīgā attīstība netiek ietekmēta.
Zīdaiņiem var rasties elpošanas traucējumi un nespēja ēst. Iedzimtas kontraktūras, dažādas smaguma pakāpes, sākot no vienkāršas kāju pēdas līdz ģeneralizētai artrogripozei (daudzveidīga iedzimta kustību aparāta patoloģija, kas izpaužas ar daudzām locītavu kontraktūrām, muskuļu iztukšošanos un bojājumiem muguras smadzenes), rodas apmēram 10% jaundzimušo ar smagiem bojājumiem. Zīdaiņi guļ nepiespiestā vardes stāvoklī, samazinās spontāna motora aktivitāte, bērni nespēj pārvarēt ekstremitāšu smagumu un labi netur galvu.
Parasti, izņemot ļoti retus, smagus gadījumus, SMA slimnīcā netiek diagnosticēta. Bērns tiek izvadīts mājās vesels, un, kad vecāki pamana zemu muskuļu tonusu, viņi tam vai nu nepiešķir nopietnu nozīmi, ja nezina, kā veselīgam mazulim jāpārvietojas, vai arī nomierinās ar klīnikas ārsta padomu. : "Neuztraucieties, viss attīstās noteiktajā laikā, tas joprojām pieaugs un darbosies." Vecāki, iespējams, nesaprot visu problēmu nopietnību, ārsts to vajadzētu redzēt, bet diemžēl poliklīnikās viņi labi nezina SMA simptomus.
Šiem bērniem ir pirmās slimības pazīmes pirms 6 mēnešu vecuma. Tas nozīmē, ka viņi paši neiegūst iemaņas sēdēt, rāpot vai staigāt. Rezultātā šo bērnu kustības amplitūda ir ļoti maza. Tajā pašā laikā SMA neietekmē kognitīvo sfēru, bērni visu saprot un jutība netiek ietekmēta. Ja jūs rīkojaties ar viņiem tāpat kā ar parastajiem bērniem - spēlējaties, lasāt, kolekcionējat piramīdas -, tad šie bērni attīstās garīgi un psiholoģiski absolūti normāli, un visas attīstības kavēšanās, ko viņiem var uzlikt neirologi, ir saistīta ar traucētu kustību aktivitāti un apstākļiem, kādos viņi atrodas. .
Vairāk nekā 2/3 bērnu ar šo slimību mirst pirms 2 gadu vecuma, daudzos gadījumos nāve iestājas agrā zīdaiņa vecumā elpošanas muskuļu bojājumu un dažādu plaušu komplikāciju dēļ.
CMA2
Ar II tipa mugurkaula muskuļu atrofiju slimība vispirms parādās nedaudz vēlāk (pirmajos 1,5 bērna dzīves gados), un to raksturo lēnāka gaita. Galvenais simptoms ir bērna nespēja piecelties.
Bērni ar SMA2 parasti spēj sūkāt un norīt, un agrā zīdaiņa vecumā elpošanas funkcija netiek traucēta. Deguna balss tonis un rīšanas traucējumi parādās vecākiem. Neskatoties uz progresējošu muskuļu vājumu, daudzi no viņiem izdzīvo līdz skolai un vecāki, lai arī vēlākos posmos slimība ir smagi invalīds, un bērniem ir nepieciešami invalīdu ratiņi. Parastie ratiņi viņiem neder, viņiem ir nepieciešami papildu balsti, pieturvietas, īpašas ierīces, kas optimāli fiksē ķermeņa stāvokli.
Daudziem pacientiem ar ilgu paredzamo dzīves ilgumu viena no galvenajām slimības komplikācijām ir skolioze un kontraktūras, kas attīstās ļoti ātri. Pat gultā gulošiem bērniem skolioze attīstās diezgan ievērojami, mugurkaula izliekums rodas nevis no stresa, bet gan no vājuma.
Bērni ar SMA2 kādu laiku ir pietiekami stabili, un vecāki var saglabāt prasmju kopumu, kuru šie bērni jau ir ieguvuši.
Pastāv termins "slimības plato", kas nozīmē noteiktu periodu, kurā bērni ir pietiekami stabili, un nav būtiskas stāvokļa pasliktināšanās, slimības progresēšana. Tas var izskatīties šādi: bērni, tāpat kā visi parastie bērni, iegūst prasmes, sākoties slimībai, viņi pārstāj attīstīties, un tad viņu stāvokļa regresija var notikt ļoti lēni vai, gluži pretēji, ļoti ātri, katram bērnam savā veidā. Vai arī tā: bērni uz noteiktu laiku iegūst prasmes, tad notiek kāds notikums vai slimība, dažas prasmes tiek zaudētas, un tad iestājas diezgan garš "plato", kura laikā var būt pat nelieli uzlabojumi, bet pēc tam notiek neizbēgama pasliktināšanās. . Slimības progresēšanas ātrums, "plato" ilgums (vai tā trūkums) un turpmākā pasliktināšanās ir ļoti individuāla.
SMA3
Kugelberg-Welander slimība- visvieglākā mugurkaula muskuļu atrofijas forma (III tipa SMA). Tas sākas vecumā no 1,5 līdz 17 gadiem. Ar šādu slimību cilvēki dzīvo ilgāk, progresēšana ir lēna. Muskuļu atrofija sākas kājās un pēc tam izplatās rokās. Zīdaiņa vecumā klīniskās izpausmes slimības var nebūt. Proksimālajās ekstremitātēs, īpaši muskuļos, attīstās progresējošs vājums plecu josta... Pacienti saglabā spēju staigāt patstāvīgi. Bulbaru muskuļu vājuma simptomi ir reti. Aptuveni 25% pacientu ar šo SMA formu ir vairāk muskuļu hipertrofijas nekā muskuļu atrofija; tāpēc var tikt nepareizi diagnosticēta muskuļu distrofija. Pacienti var dzīvot līdz pilngadībai.
SMA šajā lielajā bērnu grupā tiek atklāta vecumā virs pusotra gada, t.i. bērns parasti var staigāt patstāvīgi. Slimība izpaužas tik individuāli, ka diagnozi var noteikt pusotra gada laikā, un varbūt pat deviņu gadu vecumā. Atkarībā no tā būs dažādas prognozes gan par dzīves ilgumu, gan kvalitāti.
Bērniem ar SMA3 paredzamais dzīves ilgums ir gandrīz vienāds, viņi dzīvo līdz 30 un 40 gadiem. Visi viņu simptomi neprogresē tik ātri, taču šiem bērniem ir jāpiešķir arī rehabilitācijas nodarbības un fizikālās terapijas nodarbības.
Tagad ir diezgan liels skaits pieaugušo pacientu ar SMA, un tas nav ceturtais veids, tie ir bērni ar SMA2 un SMA3, kuri ir izauguši. Viņiem ir lielas problēmas, kas nav saistītas ar kustību un elpošanu. Neatkarīgi no tā, cik ilgi viņi dzīvo, 20 vai 30 gadus, viņi jau ir vīlušies medicīnā, jo neviens nezina, ko ar viņiem darīt. Katra bērna māte ar SMA un ikviens pieaugušais pacients ar SMA var pastāstīt par to pašu stāstu par tikšanos ar ārstu: “Nenāc pie manis, mēs nezinām, ko ar tevi darīt”; "Jūs vēl neesat miris, ah, wow." Patiešām, ārsti nezina, ko darīt ar šiem pacientiem, un saka: "Nu, tas, ko jūs vēlaties no manis, netiek ārstēts, tomēr, godīgi sakot, es nezinu, ko ar jums darīt." Neviens ar viņiem nenodarbojas, pēc 18 gadiem viņi nezina, ko ar viņiem darīt.
Šie pacienti (bieži vien neredzami pacienti, paliekot mājās) netic medicīniskā palīdzība jo viņi visu mūžu ir bijuši malā. Un viņi meklē palīdzību tikai tad, kad vairs nav iespējams ignorēt simptomus, kad stipras sāpes, t.i. vēlu - gandrīz uz nāves gultas. Vēl nesen nebija darba ar šo pacientu kategoriju. Tagad ir iespēja kaut kā uzlabot situāciju, vairāk palīdzības sniedz labdarības fondi, lielāka uzmanība tiek pievērsta šai problēmai.
Slimība SMA3 neattīstās ļoti ātri, pamazām notiek vispārēja ķermeņa vājināšanās un lēns iegūto prasmju zaudējums.
SMA4
Ceturtais SMA veids rodas cilvēkiem, kas vecāki par 25 gadiem. Slimība attīstās diezgan lēni, maz ietekmē vai neietekmē paredzamo dzīves ilgumu. Lietojot SMA4, var attīstīties trīce, var rasties vispārēja vājināšanās, tai skaitā muskuļu spēks... Laika gaitā SMA4 var izraisīt spēju zaudēt patstāvīgu kustību.
Šī ir ļaundabīgākā mugurkaula muskuļu atrofija, kas attīstās no dzimšanas vai pirmajos 1-1,5 bērna dzīves gados. To raksturo pieaugošās difūzās muskuļu atrofijas, ko papildina ļengana parēze, virzoties uz pilnīgu plegiju. Kā likums, Werdnig-Hoffmann amiotrofija tiek kombinēta ar kaulu deformācijām un iedzimtām malformācijām. Diagnostikas pamatā ir anamnēze, neiroloģiskā izmeklēšana, elektrofizioloģiskie un tomogrāfiskie pētījumi, DNS analīze un muskuļu audu morfoloģiskās struktūras izpēte. Ārstēšana ir slikti efektīva, kuras mērķis ir optimizēt nervu un muskuļu audu trofismu.
ICD-10
G12.0 Bērnu I tipa mugurkaula muskuļu atrofija [Werdnig-Hoffmann]

Galvenā informācija
Werdnig-Hoffmann amiotrofija ir vissmagākā no visām mugurkaula muskuļu atrofijām (SMA). Tās izplatība ir 1 gadījuma līmenī uz 6-10 tūkstošiem jaundzimušo. Katra 50. persona ir izmainītā gēna nesējs, kas izraisa slimības sākumu. Bet autosomāli recesīvā mantojuma veida dēļ bērna patoloģija izpaužas tikai tad, kad attiecīgā ģenētiskā novirze ir gan mātei, gan tēvam. Varbūtība, ka šādā situācijā būs bērns ar patoloģiju, ir 25%.
Slimībai ir vairākas formas: iedzimta, vidēja (agra bērnība) un vēlīna. Vairāki speciālisti izceļ pēdējo formu kā neatkarīgu nosoloģiju - Kugelberg-Welander amiotrofiju. Etiotropiskas un patoģenētiskas ārstēšanas neesamība, agrīna nāve, pacientu ar Werdnig-Hoffmann slimību uzraudzību izvirzīja starp visgrūtākajiem uzdevumiem, ar kuriem jāsaskaras mūsdienu neiroloģijā un pediatrijā.
Iemesli
Verdniga-Hofmana amiotrofija ir iedzimta patoloģija, ko kodē ģenētiskā aparāta sadalījums 5. hromosomas lokusa 5q13 lokusa līmenī. Gēnu, kurā notiek mutācijas, sauc par izdzīvošanas motoro neironu gēnu (SMN), gēnu, kas ir atbildīgs par motoro neironu izdzīvošanu. 95% pacientu ar Werdnig-Hoffmann slimību šī gēna telomēru kopija tiek izdzēsta. SMA smagums tieši korelē ar delēcijas vietas garumu un vienlaicīgu izmaiņu (rekombinācijas) klātbūtni H4F5, NAIP, GTF2H2 gēnos.
SMN gēna aberācijas rezultāts ir muguras smadzeņu motoro neironu nepietiekama attīstība, lokalizēta tās priekšējos ragos. Sekas ir nepietiekama muskuļu inervācija, kas noved pie to izteiktās atrofijas ar muskuļu spēka zudumu un pakāpenisku izmiršanu spēju veikt aktīvas motora darbības. Galvenās briesmas ir muskuļu vājums. krūtīs, bez kura piedalīšanās nav iespējamas kustības, kas nodrošina elpošanas funkciju. Tajā pašā laikā maņu sfēra paliek neskarta visā slimības gaitā.
Amiotrofijas simptomi
Iedzimta forma(SMA I) klīniski izpaužas pirms 6 mēnešu vecuma. Dzemdē tas var izpausties ar gausu augļa kustību. Bieži vien muskuļu hipotonija tiek atzīmēta jau no pirmajām dzīves dienām, un to papildina dziļu refleksu izzušana. Bērni vāji raud, slikti sūkā, nevar turēt galvu. Dažos gadījumos (ar vēlāku simptomu parādīšanos) bērns iemācās turēt galvu un pat sēdēt, taču uz slimības attīstības fona šīs prasmes ātri izzūd. Raksturo agrīni bulbaru traucējumi, samazināts rīkles reflekss, fascikulāra mēles raustīšanās.
Šī Werdnig-Hoffmann amiotrofija tiek apvienota ar oligofrēniju un traucējumiem osteoartikulārā aparāta veidošanā: krūškurvja deformācijas (piltuvveida un ķīļveida krūtis), mugurkaula izliekums (skolioze), locītavu kontraktūras. Daudziem pacientiem ir citas iedzimtas anomālijas: hemangiomas, hidrocefālija, kāju pēdas, gūžas displāzija, kriptorichidisms utt.
SMA I kurss ir pats ļaundabīgākais ar strauji pieaugošu elpošanas muskuļu nekustīgumu un parēzi. Pēdējais izraisa elpošanas mazspējas attīstību un progresēšanu, kas ir galvenais nāves cēlonis. Rīšanas traucējumu dēļ pārtika var tikt izmesta elpošanas traktā, attīstoties aspirācijas pneimonijai, kas var būt nāvējoša mugurkaula amiotrofijas komplikācija.
Agrās bērnības forma(SMA II) debitē pēc 6 mēnešu vecuma. Šajā periodā bērniem ir apmierinoša fiziskā un neiropsihiskā attīstība, saskaņā ar vecuma normām viņi iegūst prasmes turēt galvu, apgāzties, apsēsties un stāvēt. Bet pārliecinoši lielākajā daļā klīnisko gadījumu bērniem nav laika iemācīties staigāt. Parasti šī Werdnig-Hoffmann amiotrofija izpaužas pēc bērna pārtikas toksikoinfekcijas vai citas akūtas infekcijas slimības.
Sākotnējā periodā perifēra parēze notiek apakšējās ekstremitātēs. Tad viņi ātri izplatījās augšējās ekstremitātes un stumbra muskulatūra. Attīstās difūzā muskuļu hipotonija, un dziļi refleksi izzūd. Tiek novērotas cīpslas kontraktūras, pirkstu trīce, valodas piespiedu muskuļu kontrakcijas (fascikulācijas). Vēlākajos posmos bulbar simptomi pievienojas, progresējoši elpošanas traucējumi... Kurss ir lēnāks nekā Werdnig-Hoffman slimības iedzimtajai formai. Pacienti var dzīvot līdz 15 gadu vecumam.
Kugelberg-Welander amiotrofija(SMA III) - bērnības labvēlīgākā mugurkaula amiotrofija. Izpaužas pēc 2 gadiem, dažos gadījumos laika posmā no 15 līdz 30 gadiem. Garīgā attīstība neaizkavējas, pacienti ilgu laiku spēj pārvietoties neatkarīgi. Daži no viņiem dzīvo līdz pilnām vecumdienām, nezaudējot spēju pašapkalpošanās.
Diagnostika
Diagnostikas plānā bērnu neirologam pirmo simptomu parādīšanās vecums un to attīstības dinamika, dati neiroloģiskais statuss(pirmkārt, perifērā tipa motorisko traucējumu klātbūtne uz absolūti neskartas jutības fona), vienlaicīgu iedzimtu anomāliju un kaulu deformāciju klātbūtne. Iedzimtu Werdnig-Hoffmann amiotrofiju var diagnosticēt neonatologs. Diferenciāldiagnostika tiek veikta ar miopātijām, progresējošu Duchenne muskuļu distrofiju, amiotrofo laterālo sklerozi, syringomyelia, poliomielītu, gausa bērna sindromu, cerebrālo trieku, vielmaiņas slimībām.
Lai apstiprinātu diagnozi, tiek veikta elektroneuromiogrāfija - neiromuskulārā aparāta pētījums, kura dēļ tiek atklātas raksturīgas izmaiņas, kas izslēdz primāro muskuļu bojājuma veidu un norāda uz motora neirona patoloģiju. Bioķīmiska analīze asinis neatklāj būtisku kreatīna fosfokināzes pieaugumu, kas raksturīgs progresējošai muskuļu distrofijai. Retos gadījumos mugurkaula MRI vai CT vizualizē atrofiskas izmaiņas muguras smadzeņu priekšējos ragos, bet var izslēgt citas mugurkaula patoloģijas (hematomēlija, mielīts, cista un muguras smadzeņu audzējs).
"Werdnig-Hoffmann amiotrofijas" galīgā diagnoze tiek noteikta pēc muskuļu biopsijas un ģenētisko pētījumu datu iegūšanas. Muskuļu biopsijas morfoloģiskais pētījums atklāj muskuļu šķiedru patognomonisko saišķu atrofiju ar mainīgām miofibrilu un nemainītu muskuļu audu atrofijas zonām, atsevišķu hipertrofētu miofibrilu klātbūtni, saistaudu izaugumu laukumus. Ģenētikas speciālistu veiktā DNS analīze ietver tiešu un netiešu diagnostiku. Izmantojot tiešo metodi, ir iespējams arī diagnosticēt gēnu aberācijas heterozigotu pārvadāšanu, kas ir svarīgi ģenētiskajā konsultācijā slimām personām, kas plāno grūtniecību, brāļiem un māsām (brāļiem un māsām). Šajā gadījumā svarīga loma ir SMA lokusa gēnu skaita kvantitatīvai analīzei.
Pirmsdzemdību DNS pārbaude var samazināt bērna ar Werdnig-Hoffmann slimību iespējamību. Tomēr, lai iegūtu augļa DNS materiālu, ir jāizmanto invazīvas pirmsdzemdību diagnostikas metodes: amniocentēze, horiona biopsija, kordocentēze. Werdnig-Hoffmann amiotrofija, kas diagnosticēta dzemdē, ir norāde uz mākslīgu grūtniecības pārtraukšanu.
Werdnig-Hoffmann amiotrofijas ārstēšana
Etiopatogenētiskā terapija nav izstrādāta. Pašlaik Werdnig-Hoffmann amiotrofiju ārstē, uzlabojot perifērijas vielmaiņu nervu sistēma un muskuļu audus, lai palēninātu simptomu progresēšanu. Terapijā dažādu zāļu kombinācijas farmakoloģiskās grupas: neirometabolīti (preparāti, kuru pamatā ir cūku smadzeņu hidrolizāts, B grupas vitamīni, gamma-aminosviestskābe, piracetāms), atvieglo neiromuskulāro transmisiju (galantamīns, sanguinarīns, neostigmīns, ipidakrīns), uzlabo miofibrilu trofismu (glutamīnskābe Q10, koenzīms, L-karnitīns, metionīns), kas uzlabo asinsriti (nikotīna skābe, skopolamīns). Ieteicami fizioterapijas vingrinājumi un bērnu masāža.
Mūsdienu tehnoloģiju attīstība ir ļāvusi nedaudz atvieglot pacientu un viņu tuvinieku dzīvi, pateicoties automatizētu ratiņkrēslu un pārnēsājamu ventilatoru izmantošanai. Dažādas ortopēdiskās korekcijas metodes palīdz uzlabot pacientu mobilitāti. Tomēr galvenās SMA ārstēšanas perspektīvas ir saistītas ar ģenētikas attīstību un iespēju meklēšanu ģenētisko noviržu korekcijai, izmantojot gēnu inženierijas metodes.
Prognoze
Iedzimtajai Werdnig-Hoffmann amiotrofijai ir ārkārtīgi slikta prognoze. Ar tā izpausmi pirmajās bērna dzīves dienās viņa nāve parasti notiek pirms 6 mēnešu vecuma. Klīnikas sākumā pēc 3 dzīves mēnešiem nāve notiek vidēji līdz 2 gadu vecumam, dažreiz līdz 7-8 gadiem. Agrīnai bērnības formai raksturīga lēnāka progresēšana, bērni mirst 14-15 gadu vecumā.
Mugurkaula muskuļu atrofija(SMA) - deģeneratīvas slimības ar motoru neironu bojājumiem ar pirmsdzemdību perioda sākumu un progresējošu gaitu zīdaiņa vecumā un bērnība... Progresējošu muskuļu denervāciju daļēji kompensē reintervācija no blakus esošām motoru vienībām, tādējādi radot milzu motoru vienības, un vēlāk attīstās muskuļu šķiedru atrofija, kad galu galā patoloģiskajā procesā tiek iesaistīti reinnervējoši neironi. Centrālie motora neironi netiek ietekmēti.
Mugurkaula muskuļu atrofija(SMA) tiek klasificēti kā smagi zīdaini (Werdnig-Hoffmann slimība vai 1. tipa SMA), vēlu infantili, lēnāk progresējoši (2. tipa SMA) un pusaudži ar ilgāku gaitu (Kugelberg-Welander slimība vai 3. tipa SMA). Šīs klasifikācijas pamatā ir klīniskā izpausme, ieskaitot pacienta vecumu sākumā, muskuļu vājuma smagumu un slimības gaitu. Muskuļu biopsijā nav nošķirts 1. un 2. tips, lai gan 3. tipam raksturīgs pieaugušo, nevis zīdaiņu denervācijas-reinnervācijas modelis.
Dažiem pacientiem ir klīniska izpausmes ir starpposms starp 1. un 2. tipu vai starp 2. un 3. tipu. Mugurkaula muskuļu atrofijas (SMA) variants, Fazio-Londe slimība, ir progresējoša bulbāra paralīze, kas rodas smadzeņu stublājā izteiktāk izteiktu motoro neironu deģenerācijas rezultātā. muguras smadzenes ...
Iemesls mugurkaula(SMA) ir arestēšanas neesamība noteiktā ieprogrammētās šūnu nāves attīstības stadijā, kas ir normāla parādība iekšā embrija periods... No primitīvā neiroektoderma veidojas pārmērīga summa kustīgie neiroblasti un citi neironi tomēr tikai aptuveni 50% no tiem izdzīvo un nobriest, pārvēršoties neironos. "Papildu" šūnu dzīves cikls ir ierobežots, un tajās notiek deģeneratīvas izmaiņas.
Ja pie noteikta posmos process, kas aptur šūnu fizioloģisko nāvi, neattīstās, neironu nāve var turpināties vēlīnā augļa un pēcdzemdību periodā. Motora neirona izdzīvošanas gēns (SMN) aptur motoro neiroblastu apoptozi. Atšķirībā no vairuma gēnu, kas saglabājas evolūcijas ceļā, SMN ir unikāls zīdītāju gēns.
Mugurkaula muskuļu atrofijas (SMA) klīniskās izpausmes
Galvenais mugurkaula muskuļu atrofijas pazīmes(SMA) 1. tips ir smaga hipotensija, vispārējs muskuļu vājums, muskuļu masa, cīpslu refleksu neesamība, mēles, sejas un košļājamo muskuļu iesaistīšanās, acu un sfinkteru ārējo muskuļu funkcijas saglabāšana. Zīdaiņiem ar klīniskām slimības izpausmēm pēc piedzimšanas var rasties elpošanas traucējumi un nespēja ēst.
Iedzimtas kontraktūras, kuru smagums svārstās no vienkāršas kāju pēdas līdz vispārējai artrogripozei, rodas apmēram 10% smagi skarto jaundzimušo. Zīdaiņi guļ nepiespiestā vardes stāvoklī, samazinās spontāna motora aktivitāte, bērni nespēj pārvarēt ekstremitāšu smaguma spēku, viņi labi netur galvu. Vairāk nekā 2/3 bērnu ar šo slimību mirst pirms 2 gadu vecuma, daudzos gadījumos nāve iestājas agrā bērnībā.
Zīdaiņi ar mugurkaula muskuļu atrofiju(SMA) 2. tips parasti spēj sūkāt un norīt, un agrā zīdaiņa vecumā elpošanas funkcija netiek traucēta. Neskatoties uz progresējošu muskuļu vājumu, daudzi pacienti izdzīvo līdz skolas vecumam un vecākiem, lai gan vēlākajās slimības stadijās pastāv smaga invaliditātes pakāpe, un pacienti tiek turēti ratiņkrēslā. Deguna balss tonis un rīšanas traucējumi parādās vecākiem. Daudziem pacientiem ar ilgu paredzamo dzīves ilgumu skolioze kļūst par vienu no galvenajām slimības komplikācijām.
Kugelberg-Welander slimība- visvieglākā mugurkaula muskuļu atrofijas (SMA) forma (3. tips). Zīdaiņa vecumā slimības klīniskās izpausmes var nebūt. Proksimālajās ekstremitātēs, īpaši plecu joslas muskuļos, attīstās progresējošs vājums. Pacienti saglabā spēju staigāt patstāvīgi. Bulbaru muskuļu vājuma simptomi ir reti.
Apmēram 25% pacientu ar šo formu mugurkaula muskuļu atrofija(SMA) muskuļu hipertrofija ir izteiktāka nekā muskuļu atrofija; tāpēc var tikt nepareizi diagnosticēta muskuļu distrofija. Pacienti var dzīvot līdz pilngadībai. Fascikulācijas ir īpaša muskuļu denervācijas klīniskā pazīme. Plāniem bērniem fascikulācijas var rasties deltveida muskuļos, biceps brachii un dažreiz četrgalvu augšstilbā. Tomēr pastāvīgas piespiedu tārpiem līdzīgas kustības var maskēt ar biezu zemādas tauku audu slāni. Fascikulācijas ir labāk noteiktas valodā, kur zemādas praktiski nav saistaudi atdalot muskuļu slāni no epitēlija. Kad mēles muskuļi saraujas, piemēram, raudot vai izliekot mēli, fascikulācijas ir mazāk acīmredzamas nekā atslābinātā valodā.
Izstiepjot rokas uz priekšu bērniem ar mugurkaula muskuļu atrofija(SMA) bieži ir raksturīgs trīce pirkstos fascikulācijas un muskuļu vājuma dēļ. Šo trīci nevajadzētu jaukt ar trīci, kas saistīta ar smadzenīšu iesaistīšanos. Mialģija ir reti sastopama SMA.
Ar mugurkaula muskuļu atrofiju(SMA) sirds slimība neattīstās. Inteliģence tiek saglabāta, un bieži bērniem ar mugurkaula muskuļu atrofiju (SMA) ir augstākas intelektuālās spējas nekā viņu veselajiem vienaudžiem, jo enerģija, kuru nevar novirzīt fiziskām aktivitātēm, tiek realizēta sfērā intelektuālā attīstība... Turklāt slimības izraisīto sociālo ierobežojumu dēļ šie bērni biežāk mijiedarbojas ar pieaugušajiem, nevis ar bērniem.
Mugurkaula muskuļu atrofija (SMA) ir ģenētisks traucējums, kas ietekmē nervu sistēmas laukumu, kas atbildīgs par brīvprātīgo (skeleta) muskuļu kustības kontroli.
Lielākā daļa nervu šūnu, kas kontrolē, atrodas muguras smadzenēs, tāpēc slimības nosaukumā ir vārds "mugurkauls". Vārds "muskulis" nozīmē, ka viņi cieš, kas nesaņem signālus no šīm nervu šūnām. Nu, "atrofija" ir medicīnisks termins, kas nozīmē izšķērdēšanu vai "saraušanos", kas notiek muskuļiem, kad tie ir neaktīvi.
SMA mugurkaula smadzenēs ir nervu šūnu zudums, ko sauc par motoriskajiem neironiem, tāpēc tas tiek klasificēts kā motora neirons
SMA sākuma vecums un smagums dažādās valstīs ir ļoti atšķirīgs.
Kas izraisa SMA?
Lielākā daļa SMA gadījumu ir motoru neironu proteīnu, ko sauc par SMN (izdzīvošanas motoro neironu olbaltumvielu), deficīts
Šis proteīns, kā norāda tā nosaukums, ir būtisks normāla darbība motoneuroni. Nesenie arī liecina, ka SMN neesamība var tieši ietekmēt arī muskuļu šūnas.
Ir arī tādas SMA formas, kas nav saistītas ar SMN proteīnu.
KAS IR SMA VEIDI?
Ar SMN saistīta SMA
SMN - asociētā AGR parasti tiek iedalīta 3 kategorijās. 1. tips ir vissmagākais, ar agrāko sākumu, un 3. tips ir vismazāk smagākais, ar jaunāko sākuma vecumu. Daži eksperti arī izšķir 4. tipu vidēji smagai vai vieglai SMA ar pieaugušo sākumu.
Visi šie veidi ir saistīti ar ģenētisko sadalījumu (mutācijām) 5. hromosomā, kas ietekmē sintezētā SMN proteīna daudzumu. Augstāks olbaltumvielu līmenis samazina SMA smagumu. Plašāku informāciju par to, kā šīs mutācijas noved pie SMA, skatiet sadaļā - Vai šī ir ģimene?
Ne SMN SMA
Ir arī tādas SMA formas, kas nav saistītas ar SMN un nav 5. hromosomas mutāciju rezultāts. Lasiet vairāk par to sadaļā “Kas notiek ar cilvēkiem ar dažādas formas SMA? "
Spino-bulbaras muskuļu atrofija (SBMA)
Šāda veida SMA, saukta arī par Kenedija slimību, ir gēnu mutāciju rezultāts X hromosomā un būtiski atšķiras no ar SMN saistīto SMA tipu. Sīkāka informācija - attiecīgajā sadaļā.
KAS NOTIEK CILVĒKIEM AR SMN SAISTĪTIEM SMA?
Ar SMN saistītā SMA smagums ir atkarīgs no tā, cik agri parādās simptomi, kas savukārt korelē ar SMN olbaltumvielu daudzumu motoros neironos. Vēlāk simptomi un augstāks SMN olbaltumvielu līmenis liecina par vieglāku slimības progresēšanu.
Tomēr pašlaik lielākā daļa ekspertu šaubās par tik stingru atkarību un dod priekšroku nepārprotamas prognozes par kursa smagumu un paredzamo dzīves ilgumu, pamatojoties tikai uz debijas vecumu. Jaunākie pētījumi atbalsta šo elastību
1. SMA tips (Vērdna-Hofmaņa slimība)
Ja bērni ar SMA pirmajos dzīves mēnešos ir ļoti vāji un viņiem ir apgrūtināta elpošana, zīšana un rīšana, viņiem var būt grūti iegūt labu prognozi. Iepriekš tika uzskatīts, ka šādi bērni neizdzīvo ilgāk par diviem gadiem. Un tagad vairumā gadījumu šī prognoze izrādās patiesa.
Tomēr, izmantojot dabisko elpošanas un barošanas funkciju vietā tehnoloģiju, šādi bērni var izdzīvot vairākus gadus. Mehāniskā plaušu ventilācija (tagad šādas ierīces ir kļuvušas pārnēsājamas, atšķirībā no iepriekšējos gados izmantotajām "dzelzs plaušām" un smagajiem mehānismiem) un barošana caur zondēm tieši kuņģī, nevis kaklā, var pagarināt dzīvi
Garīgā un emocionālā attīstība un jutība SMA ir pilnīgi normāla parādība.
2. tipa SMA (starpposma SMA)
2. tipa SMA diagnoze ļauj vecākiem un bērniem plānot nākotni, neraugoties uz īsāku dzīves ilgumu. Šāda veida SMA debitē bērnībā, bet parasti pēc zīdaiņa vecuma. Daži avoti atsaucas uz 2. tipa slimību, kuras sākuma vecums ir no 6 līdz 12 mēnešiem. Citi avoti apgalvo, ka slimība tiek klasificēta kā 2. tips, ja bērns, ja viņu ievieto, var sēdēt bez atbalsta
Šāda veida SMA muskuļi, kas atrodas tuvāk stumbra centram (tā sauktie proksimālie muskuļi), parasti tiek skarti vairāk vai vismaz agrāk nekā muskuļi, kas atrodas tālāk no centra (distāli). Piemēram, augšstilbu muskuļi ir vājāki nekā kāju muskuļi.
Turklāt kājas mēdz vājināties agrāk nekā rokas. Rokas galu galā novājinās, bet joprojām paliek stiprākas, un, pat ja tās ir novājinātas, parasti saglabā spēku, kas ir pietiekams, lai rakstītu uz datora tastatūras un veiktu citas mūsdienu dzīves pamatfunkcijas.
Bērni ar 2. tipa SMA gūst lielu labumu no fizikālās terapijas un visu veidu palīgierīcēm. Palīglīdzekļi stāvēšanai un staigāšanai, piemēram, gaismas stiprinājumi (ortozes) un nodrošina lielāku mobilitāti, nekā tas bija iespējams agrāk.
Daudzi bērni atkarībā no attīstības pakāpes var darboties elektriskos ratiņkrēslos vai ar citiem transporta līdzekļiem diezgan agri, jau apmēram 3 gadu vecumā. Daudzi eksperti atzīmē, ka bērni ar SMA ir neparasti gudri un asprātīgi, un daži pētījumi atbalsta šos novērojumus.
Vislielākā bīstamība šāda veida SMA ir elpošanas muskuļu vājums. Rūpīga uzmanība elpošanas funkcijai un tūlītēja reakcija uz infekcijām ir būtiska visa mūža garumā. Jūs varat jums palīdzēt ar īpašām veselības saglabāšanas metodēm. elpošanas sistēmas ieskaitot tīrīšanu elpošanas trakts no izdalījumiem un iespējama plaušu ventilācija. Vairāk par to skat "Elpošanas muskuļu vājums"
Vēl viena nopietna šāda veida SMA problēma ir mugurkaula izliekums, parasti sānu virzienā, ko sauc par skoliozi. attīstās muskuļu vājuma dēļ, kas atbalsta mugurkaulu, kas ir elastīga kolonna. rada lielu diskomfortu, traucējot uzturēt normālu stāju un kustīgumu, kā arī nopietni pasliktina bērna (vai pieaugušā) ķermeņa izskatu. Daži pētījumi arī parāda, ka smags mugurkaula izliekums traucē elpot
Daudziem bērniem ar SMA skoliootiskais izliekums parādās dzīves sākumā un parasti tiek koriģēts ar fiksatoriem, līdz ir īstais laiks operācijai. Ķirurgi parasti mēdz gaidīt, līdz izaugsme pilnībā (vai gandrīz) apstājas, pirms ķirurģiski iztaisnot un nostiprināt mugurkaulu. Viņi arī uzrauga mazuļa elpošanas funkciju un to, cik ātri izliekums attīstās.
Pašlaik dzīves ilgums bērnībā sāktajā SMA ir atšķirīgs. Iespējama izdzīvošana līdz pusaudža vecumam un pat ilgāk.
3. tipa SMA (Kugelberg-Welander slimība vai mērena SMA)
Daži avoti 3. tipa SMA raksturo kā SMA, kas sākas pēc 18 mēnešu vecuma, bet citi dod priekšroku to saukt par SMA, kas sākas pēc tam, kad bērns ir sācis staigāt vai vismaz pats veicis piecus soļus.
Daudzi cilvēki ar šāda veida SMA spēj staigāt līdz 30. un 40. gadiem, lai gan daži zaudē spēju staigāt jau pusaudža gados.
Liela uzmanība joprojām jāpievērš elpošanai un iespējamam mugurkaula izliekumam. Šāda veida SMA parasti ir svarīgas palīgierīces, piemēram, elektriskie invalīdu krēsli, ierīces datora izmantošanai utt. Daži cilvēki tiek cauri tikai ar niedru un dažreiz saliekamiem krēsliem, un ratiņkrēslus izmanto tikai, lai pārvietotos lielos attālumos, piemēram, , lidostā vai tirdzniecības centrā.
Cilvēki ar šāda veida SMA dzīvo līdz pilngadībai un gūst panākumus akadēmiskajā vidē un strādā.
SMA 4. tips (SMA pieaugušajiem)
Šis SMA veids joprojām ir viegls. Pēc definīcijas šāda veida slimība debitē pieaugušā vecumā. Daži eksperti apvieno 3. un 4. tipu vai 2. un 3. tipu
KAS NOTIEK CILVĒKIEM AR CITĀM SMA formām?
Citi SMA veidi, kas nav saistīti ar SMN olbaltumvielu deficītu, ievērojami atšķiras pēc smaguma un visvairāk skartajiem muskuļiem. Dažās SMA formās, tāpat kā ar SMN saistītiem veidiem, visvairāk tiek ietekmēti proksimālie muskuļi, savukārt citās gluži pretēji - distālie, t.i., tie, kas vismaz sākumā atrodas vistālāk no ķermeņa centra. Agrīnās stadijās parasti tiek skartas rokas un kājas
Dažreiz pieaugušajiem ar nezināmas izcelsmes traucējumiem, kas ietekmē tikai muguras smadzeņu un smadzeņu apakšējās daļas motoros neironus, šo stāvokli sauc par progresējošu muskuļu atrofiju. Šis stāvoklis dažkārt progresē, iesaistot smadzeņu augšdaļas motoros neironus, un tad šis stāvoklis tiek definēts kā amiotrofiskā laterālā skleroze (ALS). Šī stāvokļa pārveidošana par ALS ne vienmēr notiek
Spino-bulbaras muskuļu atrofijas (SBMA) pazīmes ir aplūkotas turpmāk.
Kaut arī visas zināmās SMA formas ir ģenētiskas, tās ir dažādu gēnu defektu rezultāts, un tām ir atšķirīgi mantojuma modeļi un prognozes ģimenes plānošanā.
Ja jums vai jūsu bērnam tiek diagnosticēta SMA, bet tas nav saistīts ar SMN, jums jākonsultējas ar savu ārstu un, iespējams, ar konsultējošu ģenētiķi, lai uzzinātu vairāk par šī konkrētā SMA veida ģenētiku un prognozēm.
KĀ IR APSTRĀDĀTS?
Lielākās iespējamās problēmas ar SMA, jo īpaši tās, kas saistītas ar 5. hromosomu, aptuvenā smaguma pakāpē ir:
- elpošanas muskuļu vājums
- rīšanas muskuļu vājums
- muguras muskuļu vājums ar progresējošu mugurkaula izliekumu
- patoloģiska reakcija uz muskuļus relaksējošām zālēm
Šīs problēmas var un vajadzētu ārstēt un novērst.
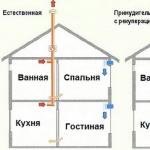
Elpošanas muskuļu vājums
Smagākajās ar SMN saistītās SMA formās un dažos citos gadījumos elpošanas muskuļu vājums ir nopietna problēma. Tas ir galvenais nāves cēlonis bērniem ar 1. un 2. tipa SMA. Kad elpošanas muskuļi ir novājināti, gaiss nevar labi cirkulēt plaušās, kas negatīvi ietekmē vispārējo veselību. Elpošanas muskuļu vājuma simptomi ir galvassāpes, grūtības gulēt naktī, pārmērīga miegainība dienā, slikta koncentrēšanās spēja, krūšu kurvja infekcijas un galu galā sirds un elpošanas mazspēja
1. tipa SMA starpribu muskuļi ir ļoti vāji, bet diafragmas muskuļi paliek diezgan spēcīgi. Šajā gadījumā bērni ir spiesti elpot vairāk ar vēdera kustību palīdzību nekā krūtīs, kas viņu ķermenim piešķir bumbieru formu.
Jaundzimušo vecākiem ar 1. tipa SMA var rasties jautājums par to, kā pagarināt bērna dzīvi pēc aptuveni diviem gadiem. Pēdējos gados pārnēsājamu un efektīvu ventilācijas ierīču pieejamība vecākiem ir devusi vairāk iespēju to izdarīt. Daži no šiem bērniem, par pārsteigumu ģimenēm un ārstiem, dzīvo daudzus gadus, dažreiz līdz pusaudža vecumam.
Dažādi ventilācijas ierīču veidi var palīdzēt pieaugušajiem un bērniem ar mazāk smagiem traucējumiem. Daudzi eksperti iesaka sākt ar neinvazīvu ventilāciju, kas parasti nozīmē, ka gaiss (parasti istabas gaiss, kas nav bagātināts ar skābekli) tiek piegādāts zem spiediena caur masku vai iemuti
Šāda veida sistēmām ir dažādas formas, un pēc vajadzības tās var izmantot vairākas stundas dienā un / vai naktī. Tos var viegli noņemt ēšanas, dzeršanas, sarunas un, kad vien iespējams, elpošanas gadījumā bez viņu palīdzības. Daži pacienti dod priekšroku negatīva spiediena (vakuuma) ventilācijas sistēmām, kas ap krūtīm vai bagāžnieku rada periodisku vakuumu, kas palīdz plaušām paplašināties un sarauties. Šīs ierīces darbojas pēc tāda paša principa kā tās, kuras tika izmantotas pirms vairākiem gadu desmitiem, t.s. "Dzelzs plaušas", bet tajā pašā laikā daudz mazāk apgrūtinošas
Bērniem un pieaugušajiem ar smagiem traucējumiem bieži ieteicams veikt ventilāciju caur traheostomiju, ķirurģisku atveri trahejā vai elpceļu. Šajā gadījumā spiediena gaiss tiek piegādāts caur traheostomijas cauruli. Parasti cilvēki var ēst, dzert un sarunāties ar caurulīti traheostomijā, lai gan šis process prasa zināmu pielāgošanos. Pastāv zināmas bažas, ka, ja bērnam ir tracheostomija, pirms viņš var runāt, tas var radīt grūtības iemācīties runāt.
Citi svarīgi aspekti elpošanas problēmu pārvaldībā SMA ir elpceļu attīrīšana, ko dažreiz veic arī ar mehāniskām ierīcēm, un pēc iespējas vairāk novēršot infekcijas.
Rīšanas muskuļu vājums
Mutes un rīkles muskuļu vājums izraisa rīšanas problēmas, īpaši vissmagākajās SMA formās. Bērniem ar 1. tipa SMA parasti ir grūtības norīt un sūkāt, kas galu galā noved pie viņu nāves. Vāja zīdīšana izraisa dehidratāciju un nepietiekamu uzturu, un norīšanas grūtības izraisa elpceļu obstrukciju pārtikas vai šķidrumu ieelpošanas (to sauc par aspirāciju) un elpceļu infekciju dēļ.
Mūsdienās var barot bērnus ar rīšanas grūtībām alternatīvas metodes, piemēram, gastrostomijas caurule (g-caurule). Mūsdienu sistēmas ir izstrādātas tā, lai caurules varētu atdalīt no vēderplēves “pogas”, kad tās nelieto. Šķidru pārtiku, kas ir viegli pieejama veikalos, ievada mēģenē, izmantojot šļirci vai barošanas sūkni. Daži cilvēki vienkārši sasmalcina parasto ēdienu.
Daži g-pipe lietotāji arī ēd un dzer parastajā veidā. Ja galvenā problēma ir košļājamo muskuļu vājums, tad ēšanas process ir grūts un laikietilpīgs, bet tajā pašā laikā ir patīkami ēst prieka un papildu uztura nolūkos, saņemot galvenās kalorijas caur g cauruli. Tomēr, ja galvenais barošanas mēģenes lietošanas iemesls ir pastāvīga pārtikas un pārtikas ieelpošana, iespējams, nav vērts mēģināt ēst un dzert normāli.
SBMA norīšanas un košļājamo muskuļu vājums rada nosmakšanas risku. Lai to noteiktu visvairāk, nepieciešams konsultēties ar speciālistu drošos veidos elpa un pārtikas konsistence. Smaga nespēka gadījumā jāapsver barošanas mēģenes izmantošana
Vāji muguras muskuļi
Bērna vecuma SMA galvenā problēma ir muguras muskuļu vājums, kas parasti atbalsta elastīgu un augstu mugurkaulu. Ja jūs tam nepievēršat uzmanību, bērnam var attīstīties skolioze (mugurkaula izliekums šķērsvirzienā) vai kifoze (izliekums garenvirzienā) vai abi. Dažreiz tas var izraisīt "kliņģera" izliekumu, padarot neiespējamu ērti sēdēt un gulēt.
Daudzi eksperti uzskata, ka smags mugurkaula izliekums negatīvi ietekmē arī elpošanas funkciju, jo izliektais mugurkauls bieži rada spiedienu uz plaušām. Tomēr smagās SMA formās ir grūti paredzēt, cik elpošanas funkcija pasliktināsies, pat ja izliekuma nav, tāpēc tās ieguldījums šajā jautājumā ir neskaidrs.
Muguras nostiprināšana ar stiprinājumiem vai stiprinājumiem, lai atbalstītu bērnu noteiktā stāvoklī, bieži tiek izmantota, mēģinot virzīt mugurkaulu tā augšanas laikā. Šādas ierīces neatrisina problēmu, lai gan tās var palēnināt izliekuma progresēšanu, kas ir vēlams
Mugurkaula izliekuma galīgais rezultāts ir ķirurģiska iejaukšanās tā fiksēšanai (tā sauktā mugurkaula saplūšana), kuru var veikt, ja bērna elpošanas stāvoklis ļauj viņam veikt šādu iejaukšanos. Parasti ārsti dod priekšroku gaidīt līdz mugurkaula augšanas beigām, lai varētu izmantot vienkāršāko ķirurģisko paņēmienu. Tomēr ne vienmēr ir iespējams gaidīt līdz augšanas beigām, jo elpošanas stāvoklis jebkurā laikā var pasliktināties. Tāpēc laiks šādai operācijai nav viegls uzdevums.
Daži vārdi par anestēziju
Bērniem un pieaugušajiem, kuriem nepieciešama operācija (piemēram, skoliozes labošanai), nepieciešami īpaši piesardzības pasākumi. Ķirurģiskajai komandai, it īpaši anesteziologam, ir labi jāpārzina SMA.
Dažreiz, īpaši SMA agrīnā stadijā, muskuļu šūnās, kas nesaņem signālus no nerviem, mēģinot sasniegt nervus, rodas noteiktas patoloģijas. Šīs novirzes var izraisīt bīstamu reakciju uz muskuļus relaksējošiem līdzekļiem, kurus parasti lieto operācijas laikā. Ja jūs zināt par šādu problēmu iespējamību, no tām var izvairīties, lietojot dažādas zāles.
Parunāsim par diētu
Daudzi cilvēki domā, vai kāda veida īpaša pārtika saviem bērniem vai viņiem pašiem var ietekmēt slimības gaitu. Tomēr pagaidām nav pierādījumu, ka kāda īpaša diēta būtu noderīga SMA.
Daudzi vecāki uzskata, ka diēta ar augstu olbaltumvielu saturu vai kāda veida īpaša uztura bagātinātāja palīdz viņu bērnu muskuļiem kļūt stiprākiem. Tomēr, lai arī nav šaubu, ka bērniem ar SMA nepieciešama laba uztura, nav pierādījumu, ka būtu nepieciešams kāds īpašs uztura veids, turklāt kaut kas varētu būt kaitīgs.
Piemēram, īpašas formulas, kas sastāv no olbaltumvielu komponentiem, ko sauc par aminoskābēm, - tā sauktās "elementārās diētas" - faktiski var radīt problēmas bērniem ar SMA, kuriem ir maz muskuļu audu. Daži eksperti norāda, ka šīs aminoskābes var paaugstināties līdz pārāk augstam līmenim asinīs, ja nav pietiekami daudz muskuļu audu, lai tos pareizi pārstrādātu.
Dažiem bērniem var noderēt biežas mazas maltītes, nevis trīs lielas maltītes dienā.
Dažiem bērniem un daudziem pieaugušajiem ar SMA ir liekais svars, iespējams, tāpēc, ka viņi fiziskās aktivitātes līmenim ēd pārāk daudz kaloriju. Ja iespējams, svars jākontrolē ārsta un dietologa vadībā, kas ir svarīgi veselībai, izskats kā arī aprūpētāju mugurām, kas ikdienā palīdz pacelt un pārvietoties
Kādi ir labākie vingrinājumi?
Lielākā daļa profesionāļu cilvēkiem ar SMA un bērnu ar SMA vecākiem saka, ka ērtas fiziskās aktivitātes, nenonākot galējībās laba ideja fiziskai un psiholoģiskai veselībai un labsajūtai
Ir svarīgi aizsargāt locītavas no nekustīguma un bojājumiem, saglabājot kustību amplitūdu (locītavu elastību), saglabājot cirkulāciju un, pats galvenais, bērniem, saglabājot pietiekamu mobilitāti, lai izpētītu apkārtējo telpu.
Īpaši noderīgas var būt nodarbības baseinā ar siltu ūdeni (30-32 grādi). Indivīdiem ar SMA nevajadzētu peldēties patstāvīgi, un ir jāveic atbilstoši drošības pasākumi
Daži eksperti izvirza jautājumu, vai ir vērts pievērst pārāk lielu uzmanību, lai pakāpeniski samazinātu motoro neironu skaitu, kuriem jāveic darbs, kuru parasti veic daudz vairāk šo šūnu. Nepieciešami pētījumi, lai noteiktu, vai šis jautājums patiešām jāņem vērā, izstrādājot vingrojumu plānu. Daži eksperti uzskata, ka pārslodze nav iespējama, bet citi uzskata, ka vingrinājumi līdz spēku izsīkumam var pirms laika "sadedzināt" atlikušos motoros neironus. Tāpēc šķiet lietderīgi iesaistīties fiziskie vingrinājumi piesardzīgi un apturiet tos pirms izsmelšanas
Fiziskās un ergoterapijas programmas var palīdzēt gan bērniem, gan pieaugušajiem iemācīties pēc iespējas labāk izmantot savu muskuļu darbību un atrast to visvairāk efektīvi veidi veicot ikdienas aktivitātes
Dažādas mūsdienās esošās palīgierīces pat mazākajiem bērniem var palīdzēt apgūt apkārtējo pasauli. Vertikālisti, staigulīši, dažāda veida elektriskie un manuālie ratiņi, ortozes var palīdzēt stāvot un pārvietojoties.
Dažas ģimenes pat izstrādā un veic savus pielāgojumus ar īpašām iezīmēm, piemēram, ar regulējamu augstumu, kas ļauj bērniem gan pārvietoties grīdas līmenī, gan sēdēt uz galda.
Īpašas ierīces rakstīšanai, zīmēšanai, datora vai tālruņa izmantošanai un elektroniskas sistēmas vides uzraudzībai (piemēram, temperatūra, apgaismojums, televizora vadība utt.) Var būt noderīgas gan pieaugušajiem, gan bērniem ar vāju SMA.
KĀ SMA tiek diagnosticēta?
Pirmais solis neiromuskulārās slimības diagnosticēšanā ir ārsta kabineta pārbaude un ģimenes anamnēze, lai atšķirtu SMA no līdzīgiem apstākļiem (piemēram, muskuļu distrofijas).
Ārsts var noteikt asins analīzi fermentam, ko sauc par kreatīnkināzi (CK). Šis ferments izplūst no muskuļiem, kad tie ir bojāti. Šis ir nespecifisks tests, jo paaugstināts CPK līmenis ir sastopams daudzās neiromuskulārās slimībās, tomēr to bieži lieto. Paaugstināts līmenis Pats CPK ir nekaitīgs, tas ir tikai muskuļu bojājumu rādītājs
Ģenētiskajai pārbaudei ir nepieciešams tikai asins paraugs. Tomēr tas ir svarīgi visai ģimenei. - Vai šī ir ģimene?)
Lai gan ģenētiskie testi ar SMN saistītām formām un SMA ir viegli pieejami, retāku SMA formu testi galvenokārt ir pieejami tikai ar pētījumu palīdzību. Tomēr šādas pārbaudes ātri uzlabojas un izplatās, uzlabojoties zināšanām un tehnoloģijām. Tuvākajā nākotnē, visticamāk, varētu izmantot ļoti detalizētus ar SMN saistītā SMA ģenētiskos testus, lai prognozētu slimības gaitu daudz precīzāk, nekā tas šobrīd ir iespējams.
Kad kādam no ģimenes ir diagnosticēti ģenētiski traucējumi vai kad viņi tiek pārbaudīti, ieteicams apmeklēt ģenētisko konsultantu.
SMA un SMA vecuma sadalījums un simptomi ar pieaugušo sākumu pārklājas ar tiem, kas slimo ar citu motoro neironu slimību, amiotrofo laterālo sklerozi (ALS), un dažreiz tiek sajaukti diagnozes sākumā. Ir ļoti svarīgi noteikt, kāda veida slimība jums vai jūsu ģimenes loceklim ir, jo ALS ir daudz smagāka un strauji progresējoša slimība nekā SMA un SMA.
Retos gadījumos cilvēkiem, kuriem ir aizdomas par SMA, tiek veikta muskuļu biopsija - ķirurģiski noņem nelielu muskuļu audu gabalu un pēc tam to pārbauda mikroskopā.
Var pasūtīt arī citus testus, piemēram, ātruma mērīšanu nervu vadīšana(ātrums, kādā signāli virzās gar nerviem) un muskuļu elektriskā aktivitāte. Pēdējo sauc par elektromiogrammu vai EMG. Nervu vadīšanas ātruma pārbaude tiek uztverta kā gaismas elektrošoki, un EMG ir nepieciešams ievadīt muskuļos īsas adatas.
KAS IR SPINO-BULBARA MUSKUĻU ATROFIJA?
Spino-bulbaru muskuļu atrofiju vai SBMA dažreiz sauc par Kenedija slimību, jo to pirmo reizi 1968. gadā aprakstīja Viljams Kenedijs. Dažreiz to sauc arī par bulbospinālo muskuļu atrofiju.
Šī slimība ir mugurkaula muskuļu atrofijas variants. Īpašības vārds "bulbar" slimības vārdā ir saistīts ar sīpolu struktūru smadzeņu apakšējā daļā, kurā ir nervu šūnas, kas kontrolē sejas, mutes un rīkles muskuļus.
Roku un kāju muskuļu vājums, īpaši tas, kas atrodas tuvāk ķermeņa centram (proksimālais), ir saistīts arī ar SBMA. Tiek novēroti arī raustīšanās vai muskuļu krampji
Kas notiek ar cilvēkiem ar SBMA?
SBMA galvenokārt skar vīriešus un parasti sākas 30-50 gadu vecumā, lai gan simptomi var parādīties gan pusaudžiem, kas jaunāki par 15, gan vīriešiem, kas vecāki par 60 gadiem. Dažām sievietēm ar šo stāvokli parasti ir viegli simptomi.
SBMA atšķiras no citiem novēlota SMA veidiem. Atšķirībā no vairuma SMA "pieaugušo" formu, SMA ievērojami ietekmē sīpolu muskuļus, kā rezultātā rodas grūtības runāt, košļāt un norīt. Rīšanas muskuļu vājums var izraisīt aizrīšanos, ēdot vai dzerot, vai pārtiku un ūdeni ieelpojot plaušās. Šī ieelpošana var izraisīt elpceļu obstrukciju (obstrukciju) vai infekciju.
Kakla muskuļu vājums var arī apgrūtināt elpošanu miega laikā. Šajā gadījumā var palīdzēt ventilācijas ierīce, kas piegādā gaisu zem spiediena.
Var attīstīties arī sejas muskuļu vājums, kas apgrūtina smaidu vai jebkādu emociju izteikšanu, izmantojot sejas izteiksmes
Tāpat kā citu SMA “pieaugušo” formu gadījumā, SMA attīstās ekstremitāšu vājums. Šis vājums vispirms vispirms izpaužas ar grūtībām kāpt pa kāpnēm un iet lielos attālumos. Šis vājums gadu desmitiem ilgi progresē ļoti lēni. Laika gaitā, ejot nelielos attālumos, var būt nepieciešama niedre, savukārt lielos attālumos var būt nepieciešams motorollers vai ratiņkrēsls
Atšķirībā no citām SMA formām, SMA raksturo ginekomastija (krūšu palielināšanās vīriešiem), samazināta auglība un sēklinieku atrofija. Šie simptomi, kas ir kļuvuši par svarīgu pavedienu slimības cēloņa noteikšanā, ir saistīti ar nepareizu vīriešu hormonu darbību, kas pazīstami kā androgēni.
SBMA ir saistīts ar X (sk. "Vai tas ir ģimenes?")
Kāds ir galvenais SBMA cēlonis?
SBMA izraisa ģenētisks bojājums X hromosomā, kas paplašina DNS daļu, ko sauc par trinukleotīdu atkārtojumu gēnā, kurā ir instrukcijas par olbaltumvielām, kas pazīstamas kā androgēnu receptori
Normāla androgēnu receptoru funkcija palīdz šūnām apstrādāt androgēnus (vīriešu hormonus). Kad androgēnu receptors satur papildu DNS, tas kļūst ilgāks nekā nepieciešams un var kļūt viskozs. Bojātais androgēnu receptors nespēj transportēt androgēnus parastajā veidā, kas izraisa motoru neironu normālas darbības traucējumus.
Vai sievietes var iegūt SBMA?
Tā kā SBMA ir saistīts ar X, tas parasti ietekmē vīriešus. Tomēr dažos SBMA gadījumos sievietes var arī saslimt, lai gan viņu slimība ir daudz vieglāka.
Pārī savienotā X hromosoma, kas sievietēm ir, parasti ļauj viņiem izvairīties no daudziem šīs slimības aspektiem. Tomēr sievietēm (kuras arī ražo un lieto androgēnus, kaut arī daudz mazāk nekā vīrieši), kurām X hromosomā ir mutācijas androgēnu receptoru gēnā (t.i., SBMA nesēji), var būt muskuļu krampji un raustīšanās, īpaši 60 -70 vecuma diapazons. Hormonālās atšķirības starp vīriešiem un sievietēm var arī veicināt mazāk smagu SMA gaitu sievietēm.
VAI ŠĪ ĢIMENE?
Kad cilvēki uzzina, ka kādam no viņu ģimenes ir ģenētiska slimība, viņi bieži vien ir pārsteigti, kā tas ir iespējams, ja "ģimenei tas nekad nav bijis"
SMA formas, kas saistītas ar 5. hromosomu, pārmanto t.s. autosomāli recesīvs tips. Lai šāda slimība varētu izpausties, ir nepieciešami divi bojāti gēni (vai nu viens no vecākiem, vai viens no vecākiem, bet otrs tā sauktās spontānas mutācijas rezultātā), cilvēki, kuriem ir tikai viens bojāto gēnu, kas nepieciešams recesīvai slimībai, sauc par "nesēju", un tam parasti nav simptomu. Bieži vien ģimenei pat nav aizdomas, ka viens no tās locekļiem ir pārvadātājs, kamēr nav piedzimis bērns ar recesīvu slimību
Autosomas ir numurētas, izņemot X un Y hromosomas, kas nosaka dzimumu
Ja abi vecāki ir ar SMN saistītas SMA nesēji, risks saslimt ar slimu bērnu katrai grūtniecībai ir 25%. Šis risks nekādā ziņā nav atkarīgs no tā, cik daudz slimu vai veselīgu bērnu iepriekš dzimuši šajā ģimenē.
Ģenētiskā testēšana ar SMA saistītām 5. hromosomas formām ir plaši pieejama tiem, kuriem ir aizdomas par šo slimību, tostarp bērniem pirms dzimšanas. Tomēr pārbaudīt SMA pārvadāšanu ir grūtāk.
Genodiagnostika strauji uzlabojas un izplatās, taču tās rezultātus var sajaukt. Tāpēc pirms tā veikšanas labāk konsultēties ar ģenētisko konsultantu.
Atšķirībā no SMN saistītā SMA, SMA bojātais gēns atrodas X hromosomā, tāpēc šo slimību pārmanto t.s. X-veida tips
Sievietēm ir divas X hromosomas, savukārt vīriešiem ir viena X un viena Y hromosoma. Sievietes, kurām vienā no X hromosomām ir bojāts gēns, tiek uzskatītas par ar X saistītas slimības nesējām (lai gan dažreiz slimība var izpausties vieglā formā). Atšķirībā no tām vīriešiem nav otrās X hromosomas, kas varētu kompensēt gēnu bojājums, tāpēc slimība izpaužas pilnībā
Tādējādi, ja vīrietis mantos bojātu gēnu X hromosomā, slimība viņam izpaudīsies. Katram sievietes pārvadātāja dēlam ir 50% iespēja iegūt bojātu gēnu un attiecīgi slimību. Katrai meitai ir arī 50% iespēja iegūt bojātu gēnu un kļūt par nesēju
Pieejama SBMA ģenētiskā pārbaude, izmantojot asins paraugus
Plašāku informāciju par mantojuma modeļiem, ģenētisko testēšanu un konsultēšanu skat "Ģenētika un neiromuskulārās slimības"
ĀRSTĒŠANAS PĒTNIECĪBA
5. hromosomas SMA pētījuma attēls pēdējās desmitgades laikā ir ievērojami noskaidrojies.
Ar SMN saistītās SMA ģenētikā ir daži īpaši apstākļi, kas pētniekiem ir devuši dažas īpašas iejaukšanās iespējas.
Kopš 1995. gada ir zināms, ka galvenais gēns, kas nosaka cilvēka slimības attīstību, ir SMN olbaltumvielu gēns. Šis gēns pastāv divās gandrīz identiskās versijās, ko sauc par SMN1 (saukta arī par SMN-T) un (vai SMN-C)
Olbaltumvielu molekulas, kas sintezētas, pamatojoties uz SMN1 gēna norādījumiem, ir garākas nekā tās, kas sintezētas, pamatojoties uz SMN2 gēnu. Šīs garākās (pilnmetrāžas) SMN olbaltumvielu molekulas ir būtiskas, lai pareizi izdzīvotu un darbotos neironu darbība.
SMN2 gēns nodrošina dažu pilnmetrāžas olbaltumvielu sintēzi, taču ar to nepietiek. Par laimi daudziem cilvēkiem vienā vai abās 5 hromosomās ir vairākas SMN2 gēna kopijas. Šie "liekie" SMN2 gēni var nedaudz kompensēt kaitīgo ietekmi, ko rada abu SMN1 gēnu kopiju bojājumi. Parasti, jo vairāk personai ir SMN2 gēna kopiju, jo lielāka iespēja, ka slimība būs vieglāka.
Daudzas SMA ārstēšanas pētījumu stratēģijas ir balstītas uz pilna izmēra SMN proteīna sintēzes palielināšanu, pamatojoties uz SMN2 gēna norādījumiem.
Citi virzieni ir balstīti uz mazāk specifiskām metodēm, kas varētu palīdzēt motoneuronu izdzīvošanai nelabvēlīgos apstākļos.
Piemēram, tiek analizētas zāles, kuru pamatā ir neirotrofiski faktori (dabiski ķermeņa produkti, kas pozitīvi ietekmē nervu šūnas); kreatīns - viela, kas palīdz muskuļu un nervu šūnām radīt enerģiju
Izmeklē arī tādas zāles kā gabapentīns, albuterols, riluzols, hidroksiurīnviela un zāles, kas pieder histona deacetilāzes inhibitoru klasei (jo īpaši fenilbutirāts un valproāts).
Gēnu un šūnu terapija attīstībā
SBMA pētījumi galvenokārt ir vērsti uz to, lai bloķētu patoloģisku agregātu veidošanos, kas konstatēti šajā slimībā šūnās; par vīriešu hormonu aktivitātes nomākšanas veidiem, kā arī par metodēm, kā ietekmēt ģenētisko instrukciju šūnu "lasīšanas" procesu.
SMA jeb mugurkaula muskuļu atrofija ir vairāku veidu iedzimtu patoloģiju grupa, kuras rezultātā notiek daļēja vai kopējie zaudējumi motora neironi muguras smadzenēs priekšā.
Šī problēma rodas sakarā ar olbaltumvielu ražojošā SMN1 gēna DNS mutāciju - mutācijas dēļ olbaltumvielu daudzums slimiem cilvēkiem ir mazāks par normu, un tas ir garantēts motoro neironu zudums.
Priekš šī slimība ko raksturo kāju, galvas un kakla šķērssvītroto muskuļu darba pārkāpums. Pacienti nespēj brīvprātīgi pārvietoties:
- rāpot;
- staigāt;
- turiet galvu;
- norīt.
Tajā pašā laikā rokas darbojas nevainojami, ir jūtīgums un laba garīgā attīstība.
Vēsturiski fakti
1891. gadā SMA bērniem pirmo reizi aprakstīja zinātnieks Werdnig. Viņš skaidri uzrādīja muskuļu, dažādu grupu, perifēro nervu un muguras smadzeņu patomorfoloģisko izmaiņu aprakstu, savukārt viņš atzīmēja muguras smadzeņu priekšējo ragu un sakņu šūnu atrofiju.
1892. gadā Hofmans atzīmēja problēmas nosoloģisko neatkarību.
1893. gadā Vērdnigs un Hofmans iesniedza pierādījumus par muguras smadzeņu raga šūnu deģenerāciju.
1956. gadā vispārējai diskusijai tika iesniegta vēlāka SMA forma ar pozitīvākām prognozēm.
Par SMA pieejama un informatīva
Jautri fakti par mugurkaula muskuļu atrofiju:
- neskatoties uz šīs problēmas retumu, tas ir visizplatītākais ģenētiskās formas pārkāpums;
- bērnībā slimība tiek pārnesta autosomāli recesīvā līnijā;
- šis gēns tiek kartēts 5. hromosomā un identificēts 1995. gadā;
- bērnu vidū vidējais līmenis dzimstība ar šo problēmu ir 1 no 6000 - 10 000 cilvēku;
- muskuļu 1. atrofijas formas bērni 50% gadījumu nedzīvo līdz 2 gadiem;
- SMA var izpausties ne tikai piedzimstot, bet arī pusmūžā vai vecumā;
- vidēji katriem 50 cilvēkiem ir recesīvs gēns;
- bērnam, kuram ir divi doti gēni gan no mātes, gan no tēva puses, ir 25% nosliece uz sakāvi; normālai veselībai pietiek ar 1 normālu gēnu.
Klasifikācija un simptomi pēc veida atšķirībām
Ir vairāki SMA veidi.
 Slimība ir pamanāma no pirmajām dzimšanas stundām, dziļu refleksu trūkuma un smagas muskuļu hipotonijas. Osteoartikulārās deformācijas ir izteiktas. Pat šim nelielajam skaitam bērnu, kas var sēdēt un turēt galvu, šīs prasmes drīz vien regresēs.
Slimība ir pamanāma no pirmajām dzimšanas stundām, dziļu refleksu trūkuma un smagas muskuļu hipotonijas. Osteoartikulārās deformācijas ir izteiktas. Pat šim nelielajam skaitam bērnu, kas var sēdēt un turēt galvu, šīs prasmes drīz vien regresēs.
Bieži vien šādiem bērniem ir gūžas locītavas, kāju pēdu, "vistas" krūtis klātbūtne.
Prognoze: slimība strauji progresē, ļaundabīga forma. Nāve iestājas pirms 9 gadu vecuma, galvenokārt problēmu dēļ kardiovaskulārā sistēma un elpošanas mazspēja.
Slimība Dubovitsa
II tips - starpprodukts, Dubovitsa slimība, līdz 18 mēnešiem. Šādi bērni pirmos attīstības posmus jau ir izturējuši labvēlīgi, taču viņi nevarēs iemācīties staigāt, slimības attīstība ir atkarīga no elpošanas muskuļu patoloģijas pakāpes.
Parasti šīs mugurkaula muskuļu atrofijas formas simptomi parādās subakūtā kursā pēc stresa ciešanas fiziskā veselība, sākas no apakšējām ekstremitātēm un pamazām virzās uz augšējām sekcijām.
Parādās mēles fascikulācija, maza ,.
Prognoze: vieglāka ļaundabīga rakstura gaita, nāve iestājas 14 - 15 gadu vecumā.
Kugelberg-Welander slimība
III tips - mazuļu, Kugelberg-Welander slimība, no 18 līdz 30 mēnešiem. Šis tips ir mazāk letāls, bērns

Fotogrāfijā programmētājs Valērijs Spiridonovs no Krievijas - diagnosticēta mugurkaula muskuļu atrofija
stāv viens pats, bet šis process viņam ir ļoti sāpīgs, nākotnē viņš būs invalīds, bez ratiņkrēsla vai niedres viņš nevar pārvietoties.
Slimība sākas nemanāmi, zīdainis sāk staigāt ar "uzvelkamās lelles" gaitu, paklūp un krīt, kustības kļūst neskaidras. Muskuļu atrofija sākotnēji ir slikti redzama attīstīto zemādas tauku klātbūtnes dēļ.
Tomēr parādās visi tie paši simptomi, fascinējošas mēles ,. Labi attīstīti refleksi izzūd jau slimības sākuma stadijā.
Notiek locītavu un kaulu deformācija, īpaši krūšu rajonā.
Prognoze: Bērni zaudē spēju staigāt par 10 - 12 gadiem un dzīvo līdz 20 - 30 gadiem.
Slimība pieaugušā vecumā
IV tips - pieaugušais, pēc 35 gadiem. Sakarā ar cīpslu refleksu atrofiju, proksimālo muskuļu samazināšanos, cilvēks zaudē spēju pārvietoties patstāvīgi.
Prognoze: strāva neietekmē dzīves ilgumu.
Citi SMA veidi un veidi
Papildus mugurkaula muskuļu atrofijai, kas tiek pārnesta ģenētiskā līmenī, ir arī vairākas saistītas slimības:
- 1. tipa X-saistītā amiatrofija - sākas no 40-60 gadiem;
- 2 - iedzimta, ļoti agresīva forma, izraisa nāvi līdz 3 mēnešiem;
- 3 - tiek skartas visas ekstremitātes, gandrīz visos gadījumos tiek skarti zēni;
- 1. tipa distālā mugurkaula amiatrofija - kopš dzimšanas, dažreiz dzemdē, tiek ietekmētas augšējās ekstremitātes;
- 2. un 4. tips - šī slimība ir aprakstīta tikai vienā ģimenē;
- 3 - lēnām attīstās slimība;
- 5 tipisks raksturs - lēnām attīstās jaunā vecumā;
- VA un VB tipa - augšējo ekstremitāšu bojājumi;
- kāju bojājums - parādās pusaudža gados, lēnām attīstās ar vājumu ceļos;
- balss saišu bojājums - saišu paralīze pieaugušajiem;
- autosomāli dominējošā mugurkaula amiatrofija - parādās pieaugušā vecumā, un tā nepilngadīgā forma pusaudža gados;
- iedzimts raksturs - slimība jau no pirmajiem dzīves mirkļiem;
- lāpstiņas-šķiedru skats - ļoti reta apakšējo ekstremitāšu slimība ar pēdas izliekumu;
- mugurkaula smadzeņu bojājuma nepilngadīgais segmenta tips ir ļoti rets gadījums, kas izpaužas pusaudža gados ar progresu 2 - 4 gadus, pēc kura tiek ietekmētas rokas;
- Fenkela slimība - attīstās galvenokārt ap 37 gadu vecumu, bet ir gadījumi, kad tā notiek 12 gadu vecumā;
- Jokela slimība - novēlota un lēna sākšanās;
- mugurkaula amiatrofija ar mioklonisko epilepsiju - tiek skartas ekstremitātes, galvenokārt mioklonisku krampju rezultātā;
- mugurkaula forma ar iedzimtiem kaulu lūzumiem - smaga muskuļu iztukšošanās forma;
- pontocerebellar hipoplāzija - ir 8 veidi;
- asimetriska segmentālā forma - 15 - 25 gadus vecu cilvēku slimība.

Diferenciālā analīze
Prognozēšana ir iespējama, pateicoties datiem, kas iegūti, kā rezultātā:
- ģenētiskā analīze;
- klīnikas pazīmes (slimības veids, atrašanās vieta);
- pavadošie simptomi, piemēram, mēles fascikulācija;
- EKG un skeleta muskuļu biopsijas dati.
Pamatojoties uz problēmām ar iedzimtu muskuļu hipotoniju - “ļengana bērna” sindromu, amiatoniju, iedzimts labdabīga muskuļu distrofija, iedzimtība un hromosomu analīze.
Nepilngadīgais izskats atšķiras no Kugelberg-Weelander mugurkaula amiatrofijas un dažādi veidi muskuļu distrofijas.

Uzdevumi un ārstēšanas metodes
 Tā kā šī slimība galvenokārt ir saistīta ar SMN olbaltumvielu trūkumu, galvenais ārstēšanas mērķis ir paaugstināt tā līmeni. Mūsdienās aktīvi šajā virzienā tiek veikti pētījumi ASV, Itālijas, Vācijas institūtos.
Tā kā šī slimība galvenokārt ir saistīta ar SMN olbaltumvielu trūkumu, galvenais ārstēšanas mērķis ir paaugstināt tā līmeni. Mūsdienās aktīvi šajā virzienā tiek veikti pētījumi ASV, Itālijas, Vācijas institūtos.
Paldies brīvprātīgo grupai klīniskie pētījumi SMA terapija ar valproiskābi, nātrija butirānu un citiem medikamentiem. Dati par cilmes šūnu lietošanu vēl nav publiskoti.
Pacientu stāvoklim ir pozitīva ietekme:

Mūsdienās pacienta stāvokli var uzturēt un nedaudz atvieglot, bet mugurkaula muskuļu atrofija joprojām ir neārstējama.
Profilakse
Profilaktiskie pasākumi ietver pasīvās darbības - konsultācijas nākamajiem vecākiem, kuriem ir SMA gēns, par iespējamo risku, pirmsdzemdību DNS diagnostika līdz 14 grūtniecības nedēļām, koriona sēnīšu paraugu ņemšana, lai veiktu galīgo diagnozi un pieņemtu lēmumus par vecāku turpmāko rīcību.
Saistītie raksti